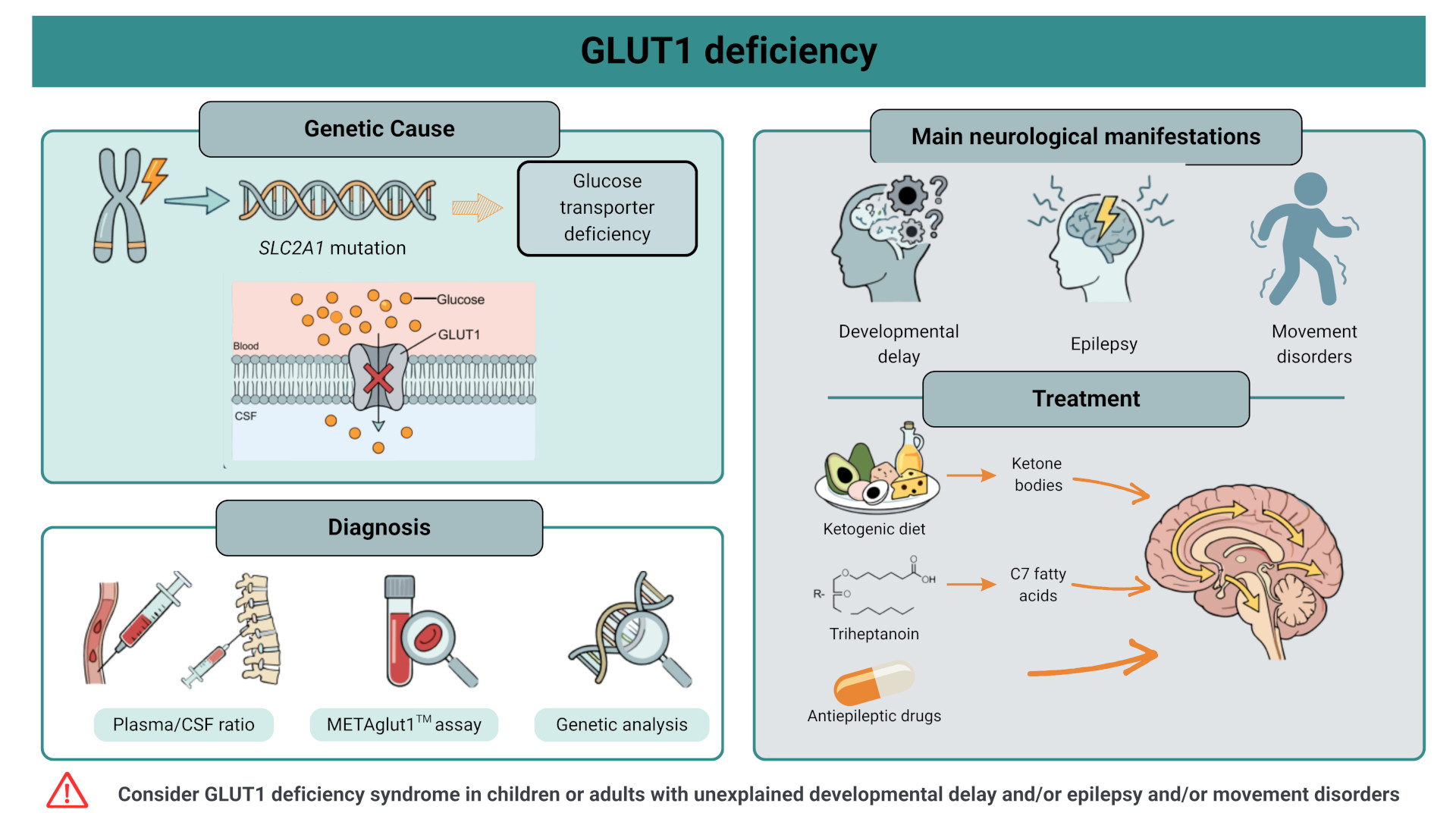
Le syndrome de déficit en GLUT1 (Glut1-DS) est une maladie génétique rare causée par des variants du gène SLC2A1, qui code pour le transporteur de glucose de type 1 (GLUT1). Ce transporteur permet le passage du glucose à travers la barrière hémato-encéphalique, fournissant ainsi au cerveau sa principale source d’énergie. Lorsque ce transport est altéré, le cerveau peut recourir aux corps cétoniques – une source alternative d’énergie produite naturellement par l’organisme en période de jeûne prolongé – pour couvrir ses besoins énergétiques.
En raison de ce déficit énergétique, les patients présentent fréquemment des manifestations neurologiques dès les premières étapes du développement cérébral. Ces symptômes sont variables et peuvent évoluer au cours du temps.
Sommaire
Cause
Le syndrome de déficit en GLUT1 est dû à des variants pathogènes du gène SLC2A1, qui code pour le transporteur de glucose de type 1. La majorité des cas résulte de variants de novo, survenant spontanément sans antécédents familiaux. Toutefois, une transmission autosomique dominante est bien décrite, avec une variabilité d’expression clinique.
Par ailleurs, le mosaïcisme est de plus en plus reconnu. Dans ces situations, le variant pathogène n’est présent que dans une proportion des cellules, en raison d’un événement post-zygotique. Cela peut conduire à des formes plus modérées ou atypiques et compliquer le diagnostic génétique, notamment lorsque le variant est présent à faible taux dans le sang.
Le diagnostic repose sur les éléments cliniques et des examens complémentaires :
- La ponction lombaire montre souvent une diminution du glucose dans le liquide céphalo-rachidien par rapport au sang (hypoglycorachie relative) ;
- Le test MetaGlut1 (notamment disponible en France) peut mettre en évidence une diminution de l’expression de GLUT1 à la surface des globules rouges ;
- L’analyse génétique confirme la présence d’un variant pathogène du gène SLC2A1.
Symptômes
Dans l’enfance :
- Retard du développement (acquisition plus lente de la marche, du langage et des apprentissages, pouvant évoluer vers un déficit intellectuel d’intensité variable)
- Épilepsie (souvent débutant dans la petite enfance, parfois résistante aux traitements, avec une tendance à s’atténuer avec l’âge)
- Microcéphalie acquise (périmètre crânien inférieur aux normes, apparaissant après la naissance
À l’adolescence et à l’âge adulte :
- Mouvements anormaux, tels que :
- Ataxie (maladresse, troubles de la coordination)
- Dystonie (contractions musculaires involontaires, postures anormales)
- Dysarthrie (troubles de l’élocution)
- Dyskinésies paroxystiques induites par l’effort ou le stress (mouvements anormaux transitoires déclenchés par l’activité physique, le jeûne ou le stress)
- Troubles cognitifs d’intensité variable (souvent légers à modérés)
- Migraines, parfois associées à des épisodes transitoires de faiblesse ou d’engourdissement d’un ou plusieurs membres
L’IRM cérébrale apporte souvent peu d’éléments d’orientation.
Traitement
Le traitement vise à fournir au cerveau une source alternative d’énergie :
- Régime cétogène : régime riche en lipides, pauvre en glucides et modérément protéique, favorisant la production de corps cétoniques utilisés comme carburant par le cerveau. Il est encadré par des diététiciens spécialisés et est particulièrement efficace sur l’épilepsie.
- Triheptanoïne : triglycéride à chaîne moyenne spécifique (à nombre impair de carbones) fournissant des intermédiaires du cycle de Krebs, constituant ainsi une source alternative d’énergie pour le cerveau. Les études cliniques suggèrent un bénéfice notamment sur les mouvements anormaux.